
By Tina Hovey, Principal Statistician, and Gillian Armstrong, Consultant Statistician, Phastar
The FDA recently published a draft guidance on Master Protocols for Drug and Biological Product Development (fda.gov) for public consultation and feedback from interested parties (such as the PSI Regulatory Special Interest Group). At Phastar, we have substantial expertise in Master Protocols therefore, our statisticians shared their expertise to review and comment on the guidance, to help shape the future direction of regulatory standards.
The FDA defines a Master Protocol as a protocol designed with multiple sub studies, which may have different objectives and involve coordinated efforts to evaluate one or more medical products in one or more diseases or conditions within the overall study structure. Essentially, it is a way of providing a common infrastructure that allows us to co-ordinate multiple sub studies, each with their own objectives.
The standard, more traditional, approach would be to conduct a series of stand-alone clinical trials each evaluating the efficacy of a single treatment in a single disease setting, supported by an independent protocol. In contrast to traditional trial designs, Master Protocols use a shared infrastructure (e.g., recruitment efforts, network of clinical sites, central facilities, central randomization system, data management systems) and share other aspects of trial design (e.g., common control arm, visit schedule, measurement procedures etc.). These features may offer efficiencies by providing a framework to test multiple drugs and/or multiple disease subpopulations in parallel under a single protocol without a need to develop new protocols for every trial. [1, 2]
There are three types of trials that can utilize a master protocol: Umbrella trials, Platform trials and Basket trials. The draft guidance focuses primarily on randomized umbrella and platform trials (not Basket trials) that are designed to provide confirmatory evidence of a drugs’ effectiveness and safety. Figure 1 illustrates the main design features of Umbrella and Platform trials.
Figure 1: Main design features of Umbrella and Platform trials
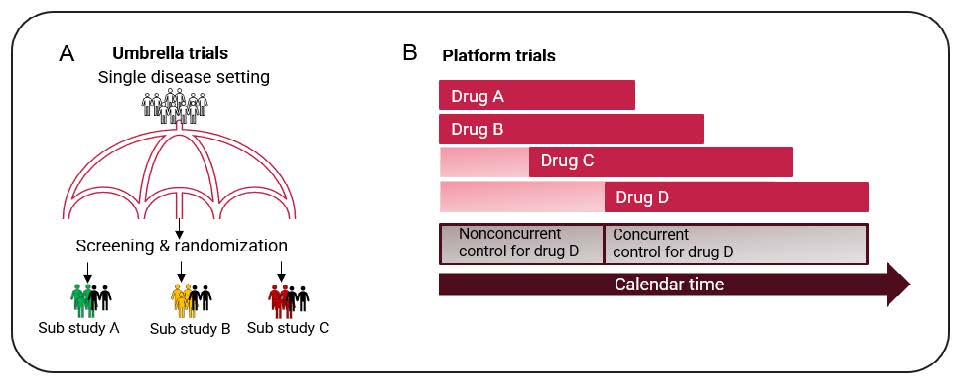
Image A illustrates an Umbrella trial where patients with common disease characteristics are randomized to one of the sub studies. Patients are further randomized to either treatment or control arm. The example above assumes there are 3 different active treatments under investigation (treatment A, B and C) and a common control. Image A adapted from: Woodcock J., LaVange L.M. Master protocols to study multiple therapies, multiple diseases, or both. N. Engl. J. Med. 2017;377(1):62–70.
Image B illustrates a Platform trial which is designed to evaluate multiple treatments for a disease in an ongoing manner, with treatments entering or leaving the platform over time. Image B adapted from: the draft FDA guidance: fda.gov
The guidance covers important design and analyses considerations for Master Protocols that are intended to contribute to a drug’s demonstration of safety and substantial evidence of effectiveness. Some important areas that resonated with us as we reviewed this guidance are:
- Optimising the randomization scheme for each drug versus control (a randomization scheme that allocates
more subjects to the control arm than each individual drug arm can increase power for each drug versus control
comparison for a given sample size). - The internal control group should generally include only concurrently randomized subjects to preserve the
integrity of the randomized comparison and valid inference. This recommendation would exclude noncurrent
controls in long running platform trials where drugs may enter the platform during or after completion of earlier
sub studies. The FDA highlights temporal shifts in patient characteristics or standard of care could cause
systemic differences between Drug A compared to a nonconcurrent control. The guidance does acknowledge
that there may be rare circumstances in which the use of nonconcurrent control data may be justified for
example in rare diseases or early phase exploratory trials, and this rationale should be discussed with the
Agency within the planning stage. - The FDA recommends obtaining informed consent upfront for the sub studies rather than using a two-stage
process of consent to the master protocol and then to treatment within a sub study. The FDA points out that
consent obtained after randomization to the sub study (i.e., subjects declining consent to their assigned sub
study) may result in subjects with different prognostic factors across the sub studies that form part of the
shared control group. This may raise concerns about the comparability of the subjects who received Drug A
compared to the shared control group. - Multiplicity, in a Master Protocol comparing multiple active drugs to control using the same primary endpoint
the FDA generally does not recommend controlling the type I error rate across these multiple comparisons (i.e.
similar to multiple independent studies that are evaluated without this control). There may be some exceptions
to this, for example when multiple doses of the same drug are being investigated. The type I error rate for other
sources of multiplicity for each individual drug/ sub study (e.g., multiple endpoints) should be controlled. - Finally blinding to treatment, the guidance covers two potential approaches to blinding treatment; a multipledummy
design where subjects are completely blinded to their assigned sub study and treatment, and a partially
blinded approach where subjects have knowledge of which sub study they have been randomized to but are
blinded to whether they receive active or control treatment (see figure 3 for a visual explanation). We agree with
the FDA comment that as the number of drugs being evaluated increases the partial blinding approach may be
more appropriate and feasible (accepting the potential for bias). Additionally, for long-running platform trials we
consider the partial blinding approach to be the only feasible approach available.
Figure 2: A Schematic to Illustrate different blinding options for an Umbrella trial.
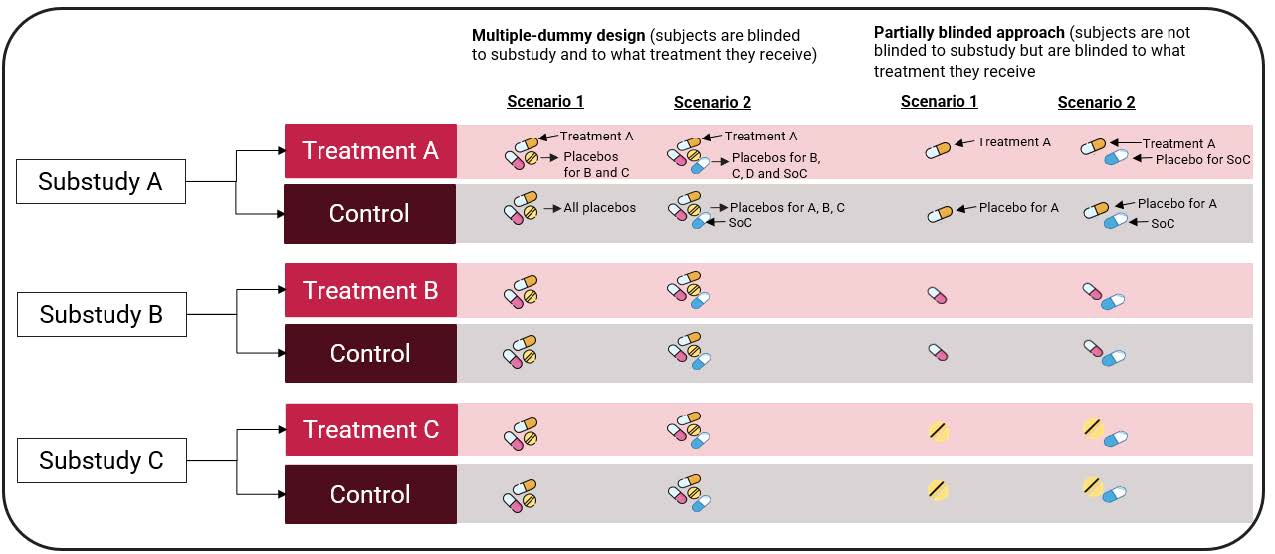
Scenario 1 assumes patients in the control arm receive a placebo, while Scenario 2 assumes control patients receive an active treatment (standard of care) instead.
SoC = Standard of Care.
The design and conduct of Master Protocols are complex and multifaceted and their role and value within the drugs clinical development program needs careful evaluation. The FDA draft guidance to industry, focussed on Master Protocols within the confirmatory setting helps this evaluation.
References
- Master Protocols for Drug and Biological Product Development Guidance for Industry, December 2023:
https://www.fda.gov/media/174976/download - Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and
Biologics Guidance for Industry, March 2022: https://www. fda.gov/media/120721/download


